M.S. Candidate: Gökçe Abay
Program: Bioinformatics
Date: 30.01.2020 / 15:30
Place: A-212
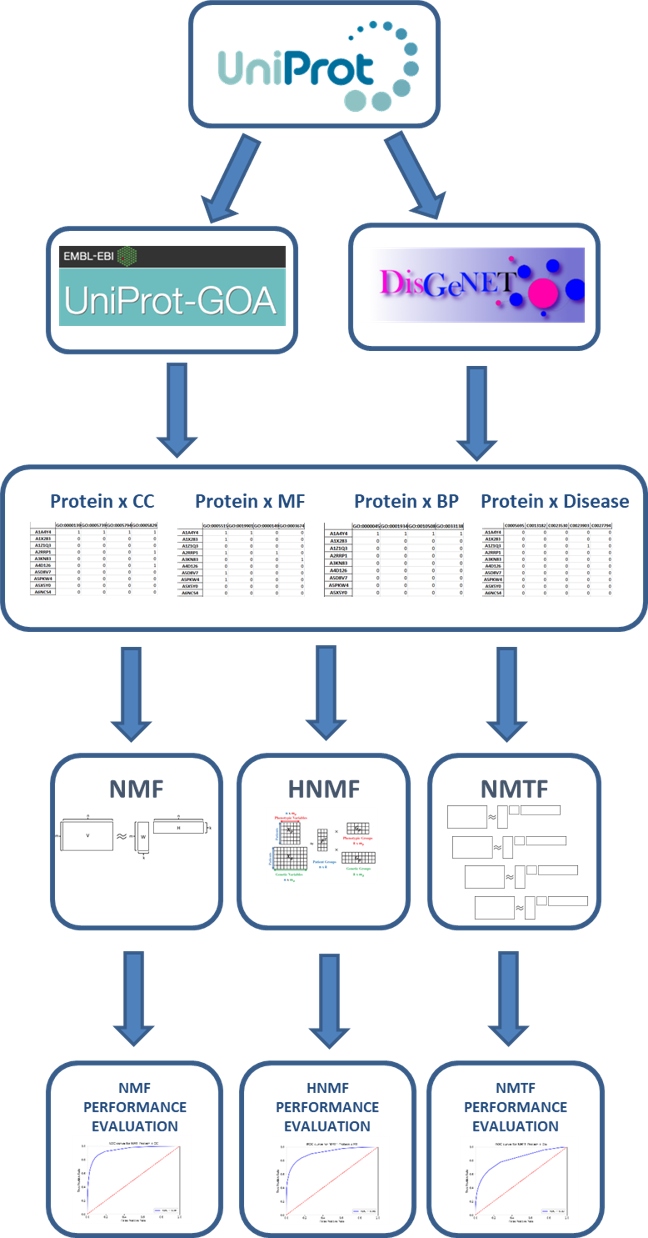
Abstract: The available molecular sequence data has increased greatly in the last decades, thanks to the new technological developments in the field of life-sciences. In order for this data to be useful to the scientific community, it should be characterized. Traditionally, this characterization is done manually, where the experimentally produced molecular data is curated and stored in the biological databases. The huge volume of the currently available data summons the need for the automatic and systematic analysis. A crucial part of this systematic analysis is data integration with the identification of the relationships between the elements from different biological data types. In this study, we propose to integrate large-scale gene/protein annotation data by using non-negative matrix factorization (NMF), which is a frequently used method for recommender systems with successful real-world applications. NMF has also been employed for uniting multi-relational data in many different fields including bioinformatics and cheminformatics. Within the purposes of this study, we first collected protein annotations such as molecular functions, biological processes, sub-cellular localizations and disease relations from different resources such as UniProt-GOA and DisGeNET, and organized them as binary relation matrices. We then applied various NMF-based algorithms to this multi-dimensional relational biomolecular sequence annotation data (i.e. genes/proteins vs. functions, genes/proteins vs. diseases, diseases vs. functions) and evaluated the results of each model in terms of their capacity to learn the intrinsic structure in relational data, via cross-validation. The results indicated that NMF has the capacity to retrieve most of the known protein annotations without using any sequence or structure-based protein features (AUROC: 0.80 – 0.94, accuracy: 0.73 – 0.87, F1-score: 0.73 – 0.89, MCC: 0.47 – 0.79). Using NMF, the ultimate aim here is to predict the unknown binary relationships between these biological entities; and to represent these entities (i.e., proteins, functions and disease entries) as informative and non-redundant quantitative feature vectors (using the low-rank feature matrices generated by the factorization process), which can be used in diverse data mining and machine learning tasks in the future, such as the automated annotations of proteins or the construction of biological knowledge graphs.