M.S. Candidate: Meriç Kınalı
Program: Bioinformatics
Date: 06.09.2019 / 10:00
Place: A-108
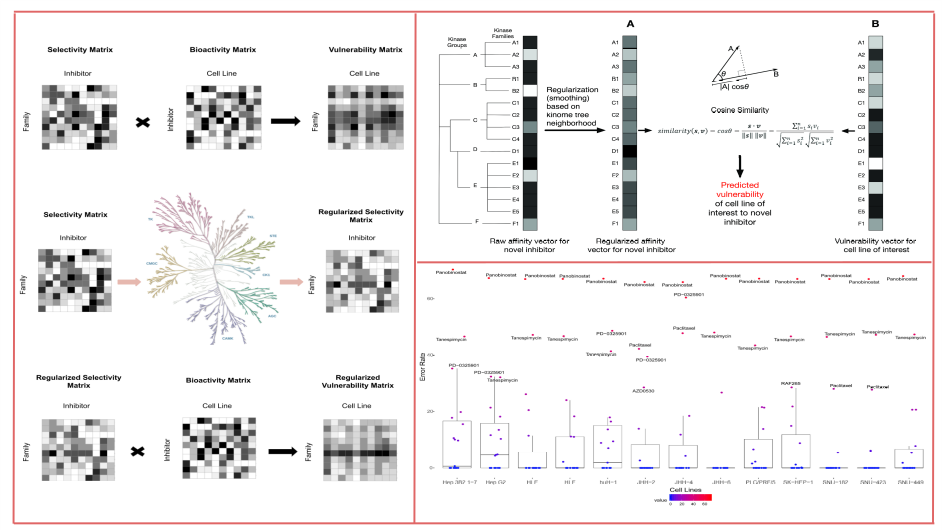
Abstract: Cancer is the second deadly disease globally. Cell signaling cascades with altered protein kinase activities induce the majority of the hallmarks in cancer such as proliferation, angiogenesis, invasion, and metastasis. The major subtype of primary liver cancer, Hepatocellular carcinoma (HCC) has limited therapeutic options. In this study, we presented a regression model, which was applied initially on cytotoxic bioactivity data obtained from HCC cells treated with 120 kinase inhibitors called CanSyL dataset. The model then extended on publicly available datasets. The model uses human kinome tree topology-based classes of protein kinases. Small-molecule kinase inhibitors can act on other pathways by “off-target” or “pathway cross-talk” effects in addition to their previously reported targets. Our objective in this study was to predict these off-target effects as potential new targets by regularizing the regression space based on the kinome tree topology. Our regression model was tested on the CanSyL dataset by applying leave-one-out cross-validation and achieved promising predictions (median RMSE between 2.5-4 %) for the kinase inhibitor vulnerability matrix based on the regularization of the human kinome tree, with no bias in the estimates. Then we scaled up our approach to the public datasets (CCLE and GDSC). Some of the kinase inhibitors were identified as outliers based on their individual RMSE. They were significantly different from the kinase inhibitor groups that they belong to, according to the Mann-Whitney U test (p<0.05). This difference in specificity suggests that outlier inhibitors are more specific inhibitors while non-outlier inhibitors are mostly general multi-kinase inhibitors.