M.S. Candidate: Elif Bozlak
Program: Bioinformatics
Date: 29.07.2019 / 11:00
Place: A-212
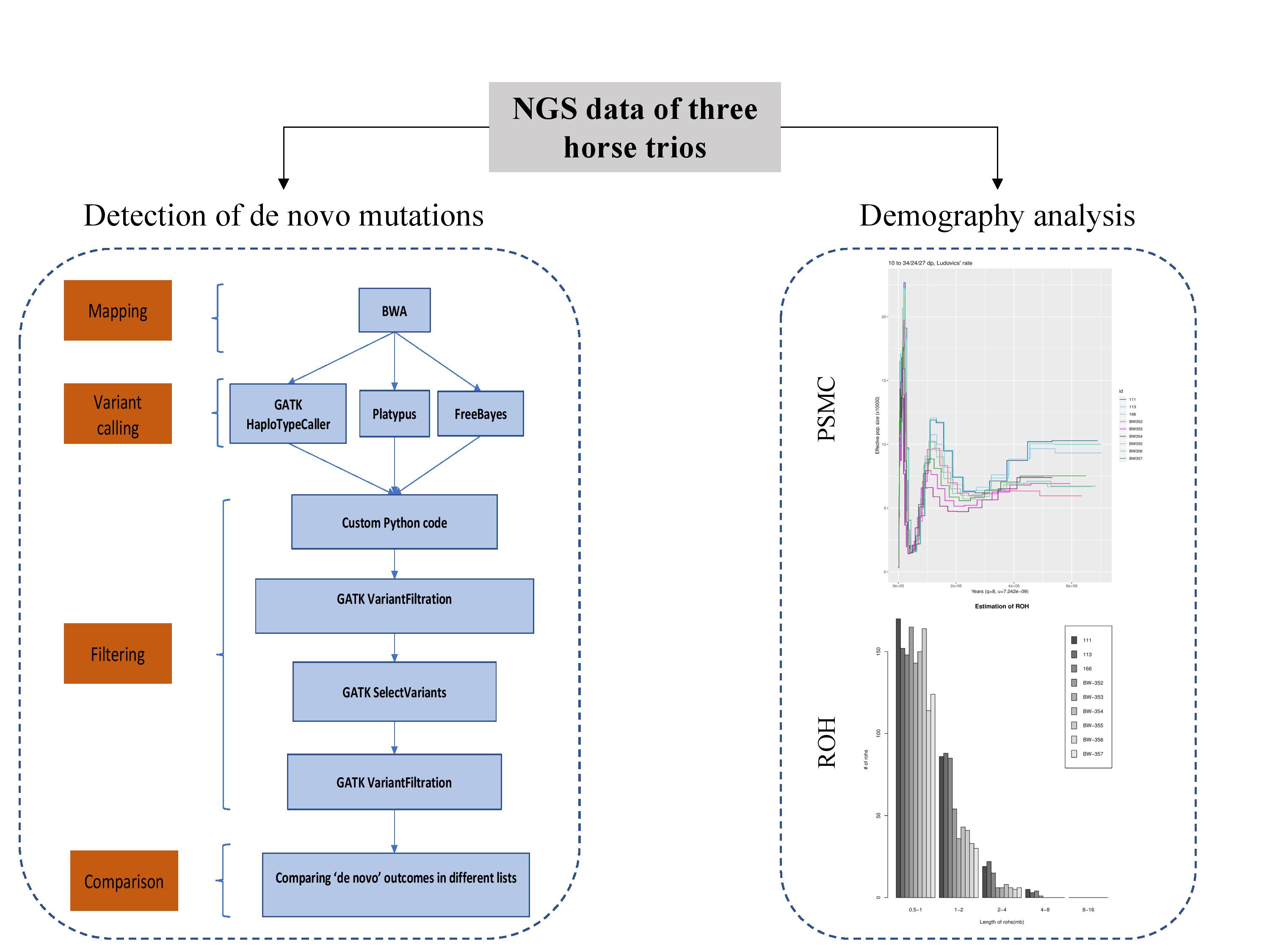
Abstract: De novo mutations are novel mutations which are found in the offspring but not the parents and do not obey the Mendelian inheritance rules. Determining how many de novo mutations occur is important for genetic studies since they help to understand the evolutionary history of populations. In this thesis, we aim to examine de novo mutations that occur within one generation in domestic horses and make estimations on horse demographic history. We used DNA-sequencing data produced by next-generation sequencing technologies from trio data of three different horse breeds: Lipizzaner, Noriker, Haflinger. After quality checks and mapping of the raw data we called genomic variants with three different variant calling algorithms. We filtered all variants depending on their qualities to detect de novo candidates and the final 50 de novo candidates were tested using Sanger resequencing. About 40% of the candidate variants could be validated. We found a higher number of true positives in highly covered Lipizzaner (n=13) data, while a lower number of true positives in the low covered Noriker (n=3) and Haflinger (n=5) data, showing the importance of sequencing coverage to detect true de novo mutations. In addition, we used the Pairwise Sequentially Markovian Coalescent (PSMC) model and performed runs of homozygosity (ROH) analyses to estimate demographic history. Both PSMC and ROH results were coherent with previous studies. All in all, we had an idea for the minimum coverage threshold and quality of whole genome sequencing data, to determine de novo mutations and to estimate population demography.