M.S. Candidate: Fatma Cankara
Program: Bioinformatics
Date: 30.01.2020 / 14:00
Place: A-212
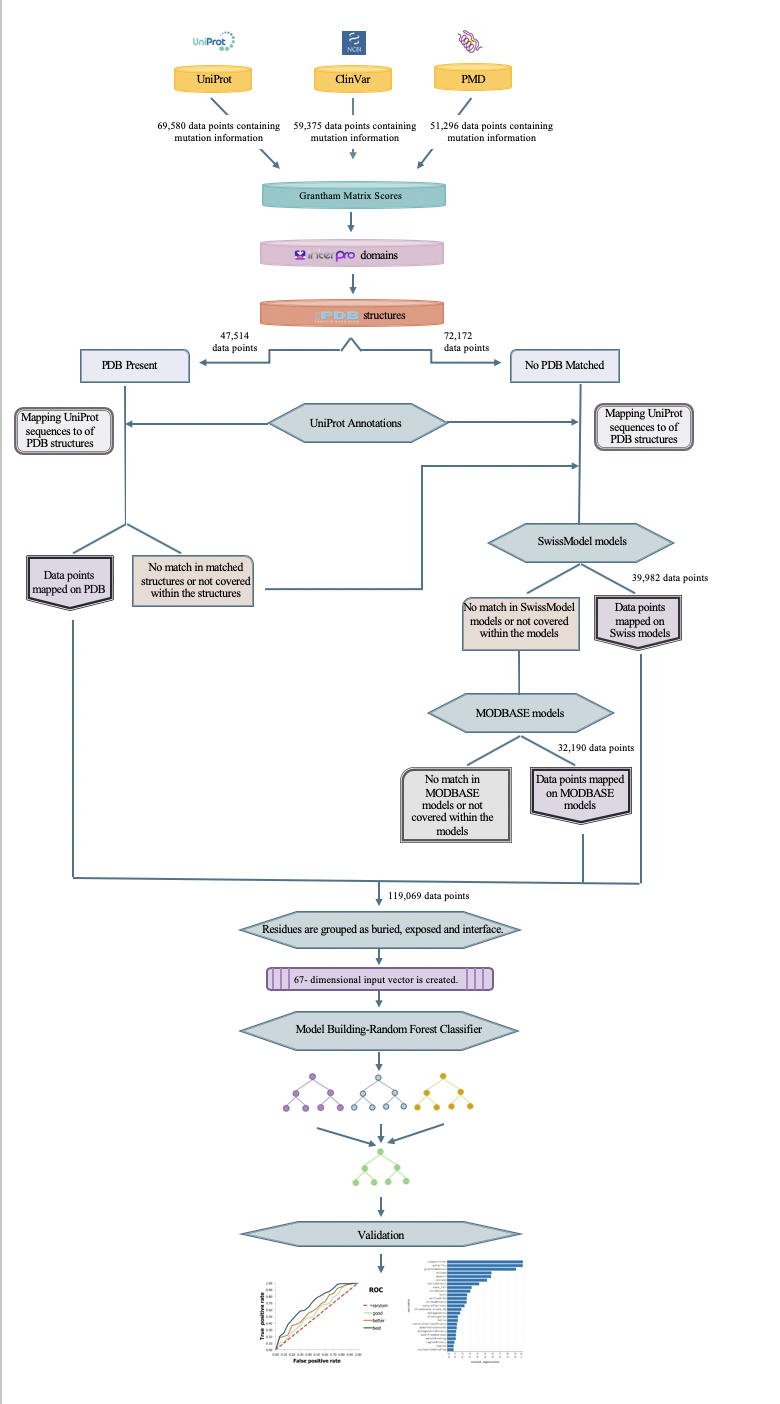
Abstract: Whole-genome and exome sequencing studies have indicated that genomic variations may cause deleterious effects on protein functionality via various mechanisms. Single nucleotide variations that alter the protein sequence, and thus, the structure and the function, namely non-synonymous SNPs (nsSNP), are associated with many genetic diseases in human. However, the current rate of manually annotating the reported nsSNPs cannot catch up with the rate of producing new sequencing data. To aid this process, automated computational approaches are being developed and applied on the unknown data. In this study, we propose a new methodology to collect and organize the information related to the effects of nsSNPs at the amino acid sequence level from various biological databases and to utilize this information in a supervised machine-learning based system to predict the function disrupting capacities of mutations with unknown consequences. For this, 109,069 annotated mutation data points (68,831 deleterious and 50,238 neutral) were collected from multiple resources such as UniProt, ClinVar and Protein Mutant Database. For each mutation data point, a feature vector was constructed using protein 3-D structure information and site-specific feature annotations in the UniProt database. The information about the spatial proximity of the reported mutations to these protein features are incorporated in the feature vectors. The system was trained with these feature vectors and their respective labels in a supervised fashion using random forest, where the ultimate aim was to construct a model that classifies unknown mutations either as deleterious or neutral. The prediction model was evaluated in detail to observe the contribution of different feature types to the prediction success. The finalized model displayed a satisfactory performance (AUROC:0.86, precision: 0.77, recall 0:90, accuracy: 0.78, F1-score: 0.83 and MCC: 0.54) on the independent test dataset. As future work, we first plan to compare the performance of our model to the widely used variant effect predictors in the literature, over benchmark datasets. Then, we will conduct a case study over interesting examples and validate our results based on literature-based information. Finally, we plan to construct a ready-to-use command line tool and shared it with the research community over an open access data repository. We hope that this system will be complementary to the well-known variant effect prediction tools in the literature and increase the performance of the state-of-the-art via its incorporation in ensemble-based tools in the future.